Hi everyone, welcome back to another case study. This post is going to talk about a case in immunodeficiency.
What is the immune system:
The immune system protects the body from infection and damage from foreign pathogens. It also plays a role in protecting us against abnormal recognition of self and the development of cancerous cells.
What is immunodeficiency:
Immunodeficiency refers to defects in the immune system, this results in gaps in the bodies defence against fighting pathogens. This can result in recurrent, severe or in some cases unusual “opportunistic” infections.
They are two types of immunodeficiency; Primary and Secondary immunodeficiency. Primary immunodeficiency is a deficiency that is present at birth due to a genetic mutation and Secondary immunodeficiency is the immune system been compromised, for example due to environmental factors or cancer treatment. Depending on the severity of the primary immunodeficiency it can present in the first few weeks of life as severe combined immunodeficiency (SCID).
Primary immunodeficiency is a deficiency that is present at birth due to a genetic mutation and Secondary immunodeficiency is the immune system been compromised due to environmental factors or cancer treatment. Depending on the severity of the primary immunodeficiency it can present in the first few weeks of life as severe combined immunodeficiency (SCID).


Case Report:
A 9 month year old baby showed to suffer from congenital neutropenia pneumonia and a reaction to BCG.
Blood was taken from the patient for an immunodeficiency (ID) test was carried out to test for the patients T&B lymphocyte subsets.
| Lymphocyte subset | Type of lymphocyte | Patients Levels | Reference ranges |
| Abs CD3 | T cells | 1056×10^6/1 | 2400-6900 |
| Abs CD4 | Helper T cells | 1040×10^6/1 | 1400-5100 |
| Abs CD8 | Cytotoxic T cells | 66 x10^6/1 | 600-2200 |
| Abs CD19 | B cells | 11 x10^6/1 | 700-2500 |
| Abs CD16/56 | NK cells | 13×10^6/1 | 100-1000 |
| Lymphosum | 1.24×10^9/1 |
The results from table 1 suggest that the patient is suffering from Lymphopenia as the results show to be below the reference ranges. The results also suggest the patient is suffering from SCID.
In the other forms of SCID you will find a complete absence of T, B and NK cells. Omenn’s syndrome is unusual in that the T&B lymphocyte subsets can look almost normal as in the patient above. This sometimes leads Omenn’s syndrome to being called a ‘leaky’ SCID!
Omenn’s syndrome
This syndrome is a rare primary immunodeficiency disorder and is classed as an autosomal recessive form of SCID.
Omenn’s syndrome is a genetically heterogeneous condition where there is a mutation in RAG-1 and RAG-2 genes resulting in partial V(D)J recombination activity and dysregulation of T and B cell functions.
The mutated RAG-1 and RAG-2 gene are involved in the coding of proteins that make up the binding part of the antibody receptor molecule, found on the surface membrane of the B Cells. These proteins are changed due to the mutations in the genes; this is called V(D)J recombination.
The proteins are unique and lead to the production of many different types of antibody receptor molecules. This allows a variety of antibodies to be selected and aids us when we suffer from a new infection or when exposed to a new allergen by allowing the antibody producing B cell to bind to the new molecule and recognise it as foreign.
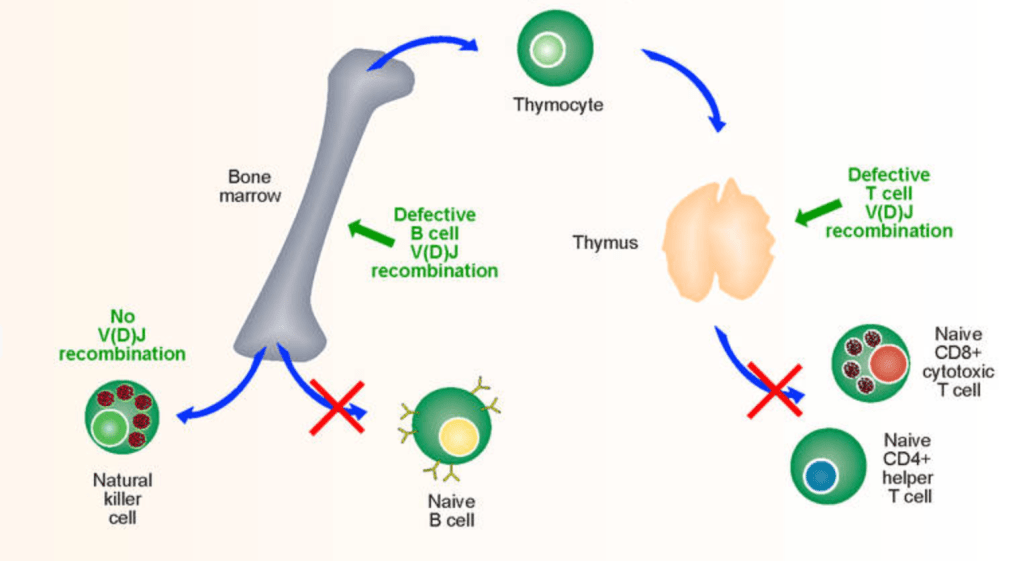
Due to the mutations on the RAG-1 and RAG-2 genes it reduces the respective proteins functions. This results in the diversity of proteins on the surface of B cells and T cells to be limited resulting in the cells to become impaired in the ability to fight of foreign invader and infections
When was the first case discovered?
The first described case was in 1965, where the clinical features of the patient were recurrent infections, skin eruptions, eosinophilia, lymphadenopathy and hepatosplenomegaly. The patient also showed to have gastrointestinal symptoms and failure to thrive.
Common Symptoms:
- Red and Peeling skin
- Chronic diarrhea
- Alopecia
- Eosinophilia
- Hepatosplenomegaly
- Lymphadenopathy
Who is more at risks in developing Omenns syndrome?
Patients that are more at risks in developing Omenn’s syndrome are people from North America, Europe and Asia.
Treatment:
Omenn’s syndrome can be treated by bone marrow transplantation or cord blood stem cell transplantation.
Special thanks to Emma (Senior BMS) and Andy (Chief BMS) for helping me with this case study.
Thanks everyone for taking your time in reading this post, leave a comment and subscribe!!

Leave a comment